
Haemoglobinometery
HSBTE IInd Semester DMLT Unit I: Haemoglobinometery Formation of hemoglobin, function, and its degradation Types of haemoglobin, Various methods of estimation with specific reference to cyanmethaemoglobin method
HAEMATOLOGY
Dr Pramila Singh
2/16/20249 min read
HSBTE IInd Semester DMLT Unit I: Haemoglobinometery: Formation of hemoglobin, function, and its degradation, Types of hemoglobin, Various methods of estimation with specific reference to cyanmethaemoglobin method.
APPLIED HAEMATOLOGY UNIT I: Haemoglobinometery
The measurement of hemoglobin content in the blood is called haemoglobinometry. It is a measurement of uncountable hemoglobin content in blood. Hemoglobin content in the blood is measured by using a hemoglobin meter. Synthesis of blood cells and blood plasma takes place in the hematopoietic system of the human body such as Red bone marrow, liver, and spleen. The synthesis of blood cells in the hematopoietic system is called hematopoiesis.
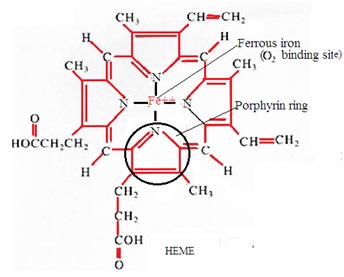
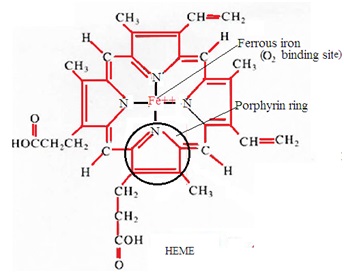
Haemoglobin is a conjugated protein consisting of haem and globin. Haem is a combination of iron and protoporphyrin. Globin is an amino acid chain. The red color of blood is due to the presence of hemoglobin in the blood. Haemoglobin carries respiratory gases in the blood.
Molecular weight of haemoglobin is 64458.
FORMATION OF HAEMOGLOBIN:
Hemoglobin synthesis takes place in immature red blood cells (RBC or erythrocytes) inside red bone marrow. Hemoglobin synthesis takes place by following a series of biochemical reactions in immature RBC inside red bone marrow. A series of biochemical reactions to synthesize hemoglobin are divided into two groups. These are a synthesis of haem and a synthesis of hemoglobin.
A. Biochemical reaction to synthesize haem:
1. A combination of Glycine and succinyl CoA (Succinyl coenzyme A) forms α-amino acid-β-keto adipic acid.
2. Enzyme amino levulinic acid synthetase converts α-ammonia acid-β-keto adipic acid to δ-amino levulinic acid (ALA).
3. Condensation of two δ-amino levulinic acid (ALA) molecules forms one molecule of porphobilinogen.
4. Condensation of four molecules of porphobilinogen forms uroporphyrinogen-I and uroporphyrinogen-III.
5. Uroporphyrinogen-I is not used in the synthesis of haem. Uroporphyrinogen-III is converted to porphyrin-I. Porphyrin-I is converted to coproporphyrinogen III. Coproporphyrinogen III to protoporphyrin IX.
6. Protoporphyrin-IX combines with ferrous iron to form haem.
B. Biochemical reaction to synthesize hemoglobin:
7. One molecule of haem combines with one molecule of haem. One hemoglobin contains four pairs of haem and globin combinations.
FUNCTIONS OF HAEMOGLOBIN:
1. Respiratory gas transportation: Haemoglobin combines with an oxygen molecule in the lungs to form oxyhemoglobin. Oxyhemoglobin transports oxygen from the lungs to cells. Haemoglobin combines with carbon dioxide in cells to form carboxyhemoglobin. Carbaminohaemoglobin transports carbon dioxide from cells to the lungs.
2. Red color to blood: Haemoglobin is pigment. The red color of blood is due to the presence of hemoglobin in RBC. The color of a single RBC is pale yellow due to the presence of hemoglobin in RBC. In the group, RBC appears as red color. This develops a red color in the blood.
3. Diagnostic tool: Break down of hemoglobin develops color in urine, stool, and bile. It helps to diagnose diseases in the human body.
4. Blood pH: Haemoglobin acts as a buffer. It helps to maintain blood pH constant.
DEGRADATION OF HAEMOGLOBIN:
Haemoglobin is present inside RBC. The average life period of RBC is 120 days. Breakdown of RBC releases hemoglobin. The breakdown of RBC is called hemolysis. Degradation of hemoglobin takes place inside reticuloendothelial cells (REC) to release globin and haem.
1. Globin is a protein part of hemoglobin. Globin is converted into amino acids. These amino acids are used to synthesize proteins inside the human body.
2. Haem is the iron part of hemoglobin. Haem combines with protein molecules to form ferritin and haemosiderin. Both are stored in the liver, spleen, and bone marrow. They are utilized in the synthesis of fresh hemoglobin.
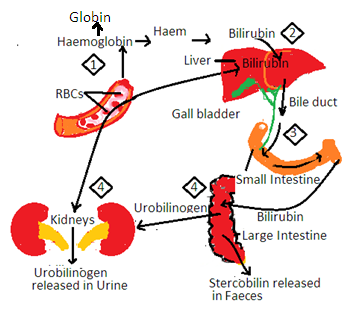
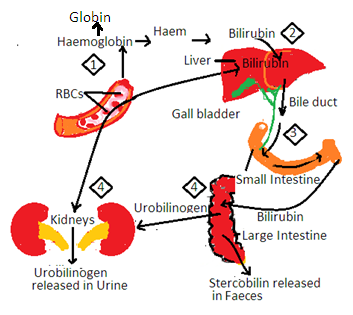
3. Some parts of haem are utilized in the synthesis of biliverdin and bilirubin in REC. Bilirubin is yellow pigment and biliverdin is green pigment. Bilirubin enters the blood.
i. Pale yellow color of blood plasma is due to the presence of bilirubin in the blood.
ii. Bilirubin enters the liver and is excreted in bile. Bile enters the small intestine with bilirubin. Intestinal bacteria convert bilirubin into urobilinogen. It develops a brown color in the stool. Some urobilinogens are reabsorbed from the intestine to enter the liverβ. It enters the blood and is excreted in the urine.
iii. The pale yellow color of urine is due to the presence of urobilinogen in urine.
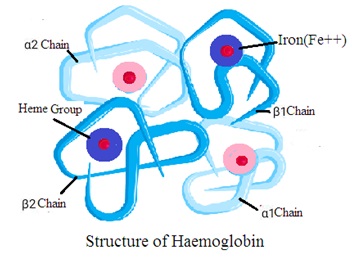
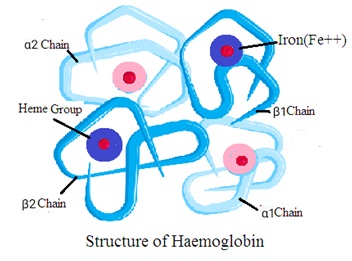
Structure of haemoglobin
Haemoglobin consists of a globin molecule attached to four heme molecules. Hemoglobin structure consists of the following two components
1. Globin: It is a protein made of four polypeptide chains. Two are alpha chains and two are beta chains. Each chain folds to form a specific structure called a globin fold.
2. Heme: It is a nonprotein ring-shaped organic molecule called porphyrin. Porphyrin tightly bonds an iron atom in the center. One hemoglobin molecule contains four heme groups. One heme group is associated with each polypeptide chain (four polypeptide chains).
Quaternary structure (Tetrameric Structure): Hemoglobin has a quaternary structure. It means hemoglobin has four globin chains and four heme groups. The four chains forms a tetrahedral structure (Tetramer Structure). Each heme group is in a pocket within the globin chain
TYPES OF HAEMOGLOBIN:
Globin is a protein part of hemoglobin. Thus globin consists of amino acids. The Globin chain has various sequences of amino acids. Following are types of hemoglobin in red blood cells depending upon sequences of amino acids in globin molecules.
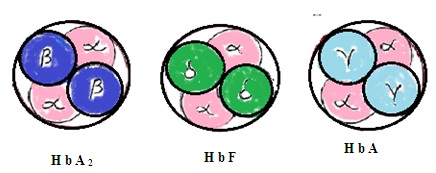
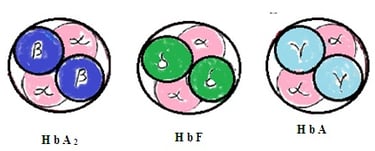
1. Haemoglobin-A (HbA): It contains 2 alpha chains of amino acids in the globin chain and 2 beta chains of amino acids in the same globin molecule. 97% of hemoglobin in RBC is HbA
Haemoglobin-A2 (HbA2): It contains 2 alpha chains of amino acids in the globin chain and 2delta chains of amino acids in the same globin chain. It constitutes 2 to 3 % of total hemoglobin in RBC.
2. Haemoglobin F (HbF2): It consists of 2 alpha chains of amino acids in a globin molecule and two gamma chains of amino acids in the same globin molecule. HbF is present in RBC during the fetus stage and just after birth in the infant. HbF is slowly replaced by HbA during 1st year of infant age. Adult human body RBC has 1 to 2% HbF. HbF is more efficient to combine with oxygen than HbA.
HbA2 HbF HbA
There are several types ’of hemoglobin depending upon their combination with respiratory gases and other chemicals. These are considered complexes of hemoglobin.
1. Oxyhaemoglobin (HbO): Haemoglobin combines with oxygen in the lungs to form a complex called oxyhemoglobin.
2. Carbaminohaemoglobin: Haemoglobin combines with carbon dioxide in cells to form a complex called carbaminohaemoglobin.
3. Carboxyhaemoglobin: Haemoglobin combined with carbon monoxide is called carboxyhemoglobin. Haemoglobin has more affinity with carbon monoxide than oxygen,
4. Methaemoglobin: Haemoglobin combines with certain drugs and their metabolites to form methemoglobin. Methaemoglobin has no affinity with oxygen. This combination oxidizes the ferrous form of iron in hemoglobin to the ferric form. This reduces hemoglobin's capacity to carry oxygen.
5. Sulphaemoglobin: Sulpha drugs combine with hemoglobin to form sulphemoglobin. It does not combine with oxygen.
Defective Haemoglobin (Haemoglobinopathies)
Symptoms that appear due to Hemoglobin synthesis disorders are called Haemoglobinopathies. These are genetic disorders of globin synthesis. Genetic disorders of haem synthesis are rare. There are three categories of Haemoglobinopathies.
1. Structural variants of hemoglobin: There are about 300 structural defective variants of hemoglobin. It is due to the substitution of single amino acids. The most common are HbS, HbC, HbD, and HbE.
2. Defective hemoglobin due to failure to form HbA. Example Thalassaemia.
3. Defective hemoglobin due to failure to switch from HbF to HbA. This disorder is called hereditary persistence of fetal hemoglobin (HPFH). These hereditary defects may be due to one normal gene and one abnormal gene or both abnormal genes.
Thalassemia
It occurs in children due to defects in genes controlling globin chain formation (alpha globin chain or beta globin chain of HbA). On this basis there are three types of Thalassemia;
1. Alpha Thalassaemia: It is due to the defective formation of alpha-globin. Alpha globin formation is controlled by four genes. Suppression of all these four genes causes alpha thalassemia. It is diagnosed by the reduced alpha/beta chain ratio.
2. Beta Thalassaemia: It is due to the defective formation of beta-globin. It is due to a defect in the HBB gene present on chromosome 11. This results in an alpha chain within red blood cells. The HBB gene has two parts. A defect in one part of the HBB gene develops Thalassaemia minor. The symptom of thalassemia minor is a small size of RBC (microcytic anemia) with less amount of hemoglobin. A defect in both parts of the HBB gene develops Thalassaemia major (Cooley’s disease). The symptom is severe hemolytic anemia.
3. Delta Thalassaemia: It is due to a defect in the HBD gene present on chromosome 11 to form a delta chain of hemoglobin..
Thalassaemia is associated with abnormal hemoglobin such as HbS, HbC, HbD, and HbE.
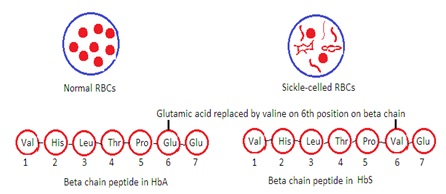
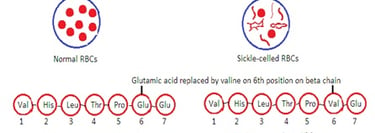
Sickle Cell Disease (HbS Disease):
It occurs due to variations in amino acid content and sequence of one of the chain pairs. Substitution of valine for glutamic acid in the 6th position of the beta chain occurs.
VARIOUS METHODS OF ESTIMATION OF HAEMOGLOBIN WITH SPECIFIC REFERENCE TO THE CYANMETHAEMOGLOBIN METHOD.
Hemoglobin content in the blood is estimated by determining hemoglobin in the blood in the form of oxyhemoglobin, carboxyhemoglobin, cyanmethaemoglobin, iron content in blood, blood color, or acid hematin. There are several methods to determine hemoglobin content in the blood. Among them, Sahli’s method and cyanmethaemoglobin method are the most common. Sahli’s method is a visual method that uses artificial standards. The cyanmethaemoglobin method is a colorimetric method that uses commercial cyanmethaemoglobin. Commercial Cyanmethaemoglobin is prepared as per the specification recommended by the International Committee for Standardisation in Haematology (ICSH).
Principal: Drabkin’s reagent and Commercial Cyanmethaemoglobin are used. Drabkin's reagent contains potassium cyanide, potassium ferricyanide, and potassium dihydrogen phosphate. The concentration of ingredients varies from manufacturer to manufacturer. It is stored in a dark bottle or dark polythene container at 2 to 8 degrees C.
Blood is mixed with Drabkin’s reagent. Hemoglobin in blood combines with potassium ferrocyanide to form methemoglobin. Methaemoglobin combines with potassium cyanide to form cyanmethaemoglobin. It develops a specific color in blood. The color intensity of blood depends upon the hemoglobin content in the blood. The color intensity of blood is compared with the color intensity of Commercial Cyanmethaemoglobin solution. It is compared by using a colorimeter to measure the optical density of both. Normally commercial Cyanmethaemoglobin solution contains 600 mg of haemoglobin per 100 mL. It will have an optical density the same as blood containing 15 gm hemoglobin per dL of blood (15 gm Hb/L).
Specimen: Venous blood of patient containing anticoagulant EDTA or double oxalate or Capillary blood of patient.
Procedure: Mix well 0.02 mL (20µL) blood with 5mL Drabkin’s solution in the cuvette. (1:250 dilution). Set aside for 5 to 10 minutes. Mark it as a “Test” solution. Measure its absorbance in a colorimeter at 540 nanometres or nm (green filter). Use Drabkin’s reagent solution as blank to set 100% T. Measure absorbance of Commercial Cyanmethaemoglobin solution as “standard” in a colorimeter at 540 nm.
Haemoglobin gm/dL = O. D. Test x 15
O. D. STD
Precautions:
1. Drabkin’s solution contains poisonous cyanide. Handle it with great care.
2. Flush Drabkin’s solution with water in the sink. Avoid mixing acid and Drabkin’s solution in the sink. This mixing releases poisonous cyanide gas.
3. Mix anticoagulated blood properly by swirling it before putting it in the cuvette.
4. Abnormal hemoglobin in the blood may develop turbidity in the cuvette after mixing blood with Drabkin’s solution.
5. Abnormal plasma protein may develop turbidity in the cuvette after mixing blood with Drabkin’s solution.
6. Do not use deteriorated Drabkin’s solution.
The normal value of hemoglobin:
· Men: 13 to 18 Hg/dL
· Women: 12 to 16.5 Hg/dL
· Infant: 11 to 13 Hg/dL
· Children: 11 to 14 Hg/dL
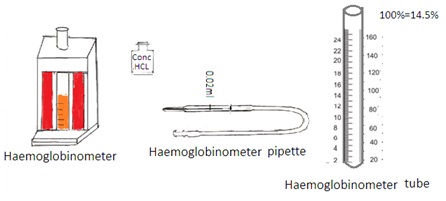
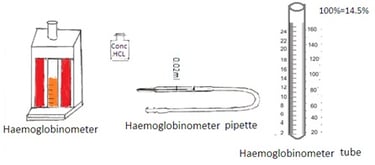
Sahli’s method (Acid Hematin Method)
Principle: It is a visual method. Hydrochloric acid reacts with hemoglobin to form hematin. Hematin is brown colored compound. The color intensity of hematin is matched visually with the brown glass present in the comparator.
Specimen: Venous blood collected in EDTA anticoagulant.
Apparatus and reagents: Sahli's graduated tube and hemoglobinometer containing comparator with glass standard. 0.1 N Hydrochloric acid. The hemoglobin pipette was marked at 20 cu mm or 0.02 ml.
Procedure: Fill 0.1 N HCL into gradutated pipette up to mark 20. Fill the blood haemoglobinometer pipette up to 20 cu mm from a finger prick. Transfer it into a tube containing acid. The blood and acid are mixed. Allow the tube to stand in the comparator on the table for 20 minutes. The brown color will develop.
Dilute the brown color by adding distilled water drop by drop. Stir after each addition. Add distilled water until the color matches with the glass plate in the comparator.
Reading: The level of the fluid in the tube indicates the gram percentage of hemoglobin in the blood.
Limitation: It is not an accurate method. Variation in result from 5% to 20%.